
Липосаркома - Liposarcoma
| Липосаркома | |
|---|---|
 | |
| Гистопатология липосаркомы, окраска H&E: - | |
| Специальность | Дерматология , общая хирургия, онкология |
| Симптомы | Уплотнение под кожей, боль, отек, нарушение функции органов |
Липосаркомы - наиболее распространенный подтип сарком мягких тканей , составляющий не менее 20% всех сарком у взрослых. Саркомы мягких тканей - это редкие новообразования с более чем 150 различными гистологическими подтипами или формами. Саркомы, которые составляют \ 1% от всех взрослых злокачественных опухолей, являются злокачественными опухолями , которые развиваются из стволовых клеток из мезенхимальных (т.е. соединительного) ткань , такие как: остеосаркомы , которые вытекают из osteoprogenitor клеток (то есть предшественник) клетки зрелых остеоцитов в костях ткани; в фибросаркомах , которые возникают из - предшественников клеток фиброцитов в соединительной ткани ; и рабдомиосаркомы, которые возникают из клеток-предшественников миоцитов в мышечных тканях . Липосаркомы возникают из предшественников lipoblasts этих адипоцитов (т.е. жировых клеток) в жировой ткани (т.е. жир) тканей . Жировая ткань распределена по всему телу, включая такие участки, как глубокие и более поверхностные слои подкожных тканей, а также в менее хирургически доступных участках, таких как забрюшинное пространство (то есть пространство за брюшной полостью ) и висцеральный жир внутри брюшной полости .
Все липосаркомы состоят по крайней мере из некоторых клеток, которые имеют сходство с жировыми клетками при исследовании их гистопатологического внешнего вида под микроскопом. Тем не менее, липосаркомы действительно есть несколько форм , основанных на различиях в их клинических проявлений (например , возраст, гендерные предпочтения, участки опухоли, признаки и симптомы ), тяготы (т.е. потенциально вторгаться местными тканями, повторялись после хирургического удаления и метастазировать в дистальных тканей), генетические аномалии , прогнозы и предпочтительные схемы лечения. Всемирная организация здравоохранения в 2020 году реклассифицировала липосаркомы на пять более или менее различные формы: 1) атипичный lipomatous опухоли / хорошо дифференцированную липосаркому; 2) дедифференцированная липосаркома; 3) миксоидная липосаркома; 4) плеоморфная липосаркома; и 5) миксоидной плеоморфная липосаркома. ( Плеоморфизм указывает на присутствие клеток, которые имеют аномальные и часто большие вариации в размере и форме и / или размере и форме их ядер.)
Хотя формы липосарком классифицируются как агрессивные и злокачественные или, в случае атипичной липоматозной опухоли / хорошо дифференцированной липосаркомы, как относительно неагрессивные и доброкачественные, все пять форм липосарком могут проникать локально, поражая близлежащие ткани и органы, возникающие в хирургически недоступные участки, прилегающие к жизненно важным органам (например, забрюшинное пространство), рецидивируют после хирургического удаления и прогрессируют до опасных для жизни заболеваний. На сегодняшний день исследования показывают, что все пять форм липосаркомы, хотя обычно поддаются лечению, по крайней мере, на начальном этапе хирургической резекцией, часто лишь незначительно реагируют на используемые в настоящее время схемы химиотерапии и лучевой терапии . Липосаркомы требуют проведения широкого круга дальнейших исследований для определения их чувствительности к различной лучевой терапии , химиотерапии и более новым схемам лечения, которые используются индивидуально и в различных комбинациях, которые включают, где это возможно, хирургическое удаление.
Формы липосарком
Липосаркомы обычно представляют собой большие опухоли (> 10 см), но могут быть практически любого размера. Они встречаются в основном у взрослых, и только 0,7% случаев возникают у лиц моложе 16 лет. У взрослых липосаркомы возникают преимущественно в среднем возрасте и старше. В очень редких случаях, встречающихся у детей и подростков, диагностируется преимущественно миксоидная форма липосаркомы.
Пять форм липосарком следует отличать не только друг от друга, но и от некоторых других опухолей мягких тканей. Этими другими опухолями наряду с некоторыми из их отличительных гистопатологических особенностей являются: 1) диспластические липомы (т.е. доброкачественные образования с участками некроза тканей и неопластическими жировыми клетками различного размера, содержащими ядра различного размера / формы ; эти неопластические клетки, в отличие от большинства неопластических клеток в липосаркомы, не сверхэкспрессии в MDM2 гена); 2) атипичные липомы веретенообразных клеток (т.е. доброкачественные опухоли с умеренно атипичными веретенообразными клетками в строме фиброзно-миксоидной формы, смешанные с вакуолизированными липобластами и адипоцитами переменного размера с атипичными ядрами; 3) плеоморфные липомы (т.е. доброкачественные опухоли, характеризующиеся гигантскими клетками). с перекрывающимися ядрами); и 4) солитарные фиброзные опухоли (т.е. опухоли, до 22% которых проявляют злокачественное поведение, состоящие из клеток веретеновидной или яйцевидной формы в коллагеновой фоновой строме, смешанных с кровеносными сосудами с характерной формой оленьего рога).
Атипичная липоматозная опухоль / высокодифференцированная липосаркома
Вместе атипичные липоматозные опухоли (ALT) и хорошо дифференцированные липосаркомы (WDL) составляют от 40% до 45% всех липосарком. Они редко, если вообще когда-либо, дают метастазы и поэтому считаются доброкачественными или предраковыми опухолями. Однако они являются местно-инвазивными и могут трансформироваться в более агрессивную и потенциально метастазирующую липосаркому, то есть в дедифференцированную липосаркому. Кроме того, хирургически удаленная атипичная липоматозная опухоль / хорошо дифференцированная липосаркома может рецидивировать в виде дедифференцированной липосаркомы.
Презентация

ALT и WDL считаются практически идентичными опухолями, за исключением того, что по определению ALT обозначают опухоли, которые развиваются в руках или ногах, в то время как WDL обозначают опухоли, которые развиваются в менее хирургически доступных местах, таких как глубокие центрально расположенные мягкие ткани забрюшинного пространства , паратестикулярная область ( т.е. область в мошонке, включая яички , семенной канатик , тунику яичка , придаток яичка и придаток яичка ), ротовую полость и глазницу . Эта терминология имеет прогностическое значение: менее 7% опухолей ALT превращаются в дедифференцированные липосаркомы в среднем за 7 лет, в то время как 17% опухолей WDL превращаются в эту более злокачественную липосаркому в среднем за 8 лет. Опухоли ALT и WDL (далее именуемые ALT / WDL) обычно представлены у людей среднего и пожилого возраста в виде медленно увеличивающихся масс, которые, как правило, больше и находятся на более продвинутой стадии, когда расположены в глубоких тканях. Эти опухоли обычно безболезненны и при поверхностном расположении легко заметны; они также могут вызывать обширный отек (то есть отек из-за локального скопления жидкости) в пораженных участках, таких как бедро (см. рисунок рядом), из-за их проникновения в кровеносные и / или лимфатические сосуды, дренирующие место опухоли. Глубоко расположенные опухоли ALT / WDL могут протекать бессимптомно, но, в зависимости от их местоположения, вызывают серьезные признаки и / или симптомы дисфункции в любом из различных органов, в которые они инфильтрируются. Эти органы включают органы, расположенные рядом с забрюшинным пространством или в нем (например, кишечник, почки и мочеточники почек ); паратестикулярная область; средостение (например, трахею и большие бронхи легких ); и голова (например, ретробульбарное пространство за глазным яблоком).
Патология

Гистопатологически опухоли ALT / WDL делятся на адипоцитарные / липомоподобные, склерозирующие и воспалительные варианты, причем адипоцитоподобные / липомоподобные являются наиболее распространенными. Адипоцитарные / липомоподобные опухоли ALT / WDL состоят из долек зрелых жировых клеток, неоднородных фиброзных перегородок (см. Соседнюю микрофотографию, окрашенную H&E ). Склерозирующие опухоли ALT / WDL, второй по распространенности вариант, развиваются в основном в забрюшинной и паратестикулярной областях; он состоит из рассеянных атипичных стромальных клеток на фоне коллагеновой (т. е. содержащей коллаген ) стромальной ткани . Редкая вакуоля отработанного lipoblasts заполнить эту ткань. Воспалительные опухоли ALT / WDL - самый редкий вариант. они чаще всего встречаются в забрюшинном пространстве и состоят из хронических воспалительных клеток, например лимфоцитов и плазматических клеток, а также случайных фолликулов, похожих на лимфатические узлы, вкрапленных по всему тканевому фону, содержащему жировые клетки .
Генетика
Неопластические клетки в опухолях ALT / WDL содержат одну или несколько дополнительных кольцевых хромосом с малыми дополнительными маркерами (sSMC) или аномальную гигантскую маркерную хромосому (т. Е. Ранее нормальную хромосому, которая стала аномальной из-за дублирования собственных частей или одной или генетический материал другой хромосомы). Эти аномальные хромосомы содержат дополнительные копии длинного плеча хромосомы 12 (также называемого плечом q ) на полосах с 13 по 15. Этот участок хромосомы 12 включает протоонкоген MDM2 (потенциально вызывающий опухоль ген при сверхэкспрессии ), расположенный на полосе 15 и CDK4 (ген, избыточная экспрессия которого способствует развитию различных опухолей), расположенный в полосе 14.1. Amplificiation (т.е. увеличена копия гена без пропорционального увеличения других генов) из этих двух генов является высоко чувствительным и специфичным показателем того, что липосаркома является либо ALT / ВЦБ или дедифференцируются липосаркома , а не любая другая липосаркомы или липомы формы. Помимо генов MDM2 и CDK4 , эта полоса 13–15 хромосомной области также содержит гены TSPAN31 и HMGA2, которые при сверхэкспрессии связаны с различными опухолями и / или раками. Было высказано предположение, что один или несколько из этих сверхэкспрессированных генов стимулируют и / или вносят вклад в развитие и / или прогрессирование опухолей ALT / WDL.
Диагностика
Диагноз опухолей ALT / WDL ставится на основании особенностей их клинических проявлений, гистопатологии и генетических данных. В частности, обнаружение в опухолевых клетках ALT / WDL сверхэкспрессированного гена MDM2 или CDK4 или наличие либо специфической ALT / WDL-ассоциированной sSMC, либо гигантской маркерной хромосомы (как определено секвенированием ДНК следующего поколения , сравнительной геномной гибридизацией и / или или высокоспециализированный цитогенетический анализ G-полос ) убедительно подтверждает диагноз ALT / WDL или дедифференцированной липосаркомы. Различия в клинической картине и гистопатологии между двумя последними формами липосаркомы обычно помогают различать их.
Лечение и прогноз
Опухоли ALT / WDL лечат радикальной хирургической резекцией для удаления всех опухолевых тканей. Однако в 30–50% случаев эти опухоли рецидивируют локально. Рецидивы чаще всего возникают в опухолях, расположенных в менее доступных местах, таких как забрюшинное пространство, средостение и семенной канатик. Эти опухоли, которые не поддаются хирургической оценке, имеют тенденцию повторяться неоднократно и в конечном итоге могут привести к смерти из-за их повреждающего воздействия на жизненно важные органы. В то время как опухоли ALT / WDL имеют очень небольшой потенциал к метастазированию , около 10% преобразуются в явно злокачественную и потенциально метастазирующую форму липосаркомы, дедифференцированную липосаркому. Среднее время этой злокачественной трансформации составляет около 7–9 лет. Кроме того, хирургически удаленный ALT / WDL может рецидивировать через переменный интервал как дедифференцированная липосаркома. Большое рандомизированное контролируемое исследование, сравнивающее лучевую терапию с последующим хирургическим вмешательством и только хирургическое вмешательство при опухолях ALT / WDL, обнаружило небольшую разницу между двумя режимами. Более мелкие исследования с использованием селективных ингибиторов белковых продуктов генов CDK4 или MDM2, участвующих в ALT / WDL, показали в лучшем случае лишь умеренные эффекты. Дальнейшие исследования с использованием этих или совершенно новых схем лечения находятся в стадии изучения. Обзорное исследование, проведенное в 2012 году, показало, что 5- и 10-летняя выживаемость людей с ALT / WDL составила 100% и 87% соответственно.
Новые методы лечения
Новые методы лечения ALT / WDL аналогичны тем, которые перечислены в разделе «Новые методы лечения» Дедифференцированной липосаркомы.
Дедифференцированная липосаркома
Дедифференцированные липосаркомы - это злокачественные опухоли, которые примерно в 10% случаев развиваются в существующей атипичной липоматозной опухоли / хорошо дифференцированной липосаркоме (ALT / WDL) или на том месте, где опухоль ALT / WPL была удалена хирургическим путем. Люди с диагнозом de novo этой опухоли могли иметь ALT / WDL, которая прогрессировала до дедифференцированной липосаркомы, но оставалась необнаруженной, потому что она развивалась бессимптомно в сильно изолированном участке, таком как забрюшинное пространство или брюшная полость. Многие клинические и генетические особенности дедифференцированных липосаркомных опухолей сходны с таковыми, обнаруженными в опухолях ALT / WDL.
Презентация
Дедифференцированные липоосаркомы (DDL) чаще всего встречаются у людей среднего и пожилого возраста, а пик заболеваемости приходится на шестой-восьмой десяток лет. В редких случаях эти опухоли развиваются у детей и подростков. Опухоли DDL чаще всего возникают в забрюшинном пространстве, но, как и ALT / WDL, могут возникать в конечностях, паратестикулярной области, средостении, голове или шее. Менее 1% всех DDL развиваются в поверхностных мягких тканях или в глазнице. При представлении DDL-опухоли обычно безболезненны, имеют большие размеры, могут медленно и прогрессивно увеличиваться в течение многих лет, и на обычных рентгеновских снимках содержат области отложения кальция (пример представлен на рис. 1 в разделе «Гистопатология липосарком»). Реже у пораженных людей есть признаки и / или симптомы из-за соударения опухоли с органом (например, боль в животе, вызванная закупоркой кишечника или обструкцией мочевыводящих путей, вызванной закупоркой уретры ). Очень редко пациенты с DDL имеют один или несколько признаков или симптомов хронического воспаления (см. Симптомы B ) и / или один из эндрокринных , неврологических , кожно-слизистых , гематологических или других тканевых паранеопластических синдромов . Признаки и симптомы хронического воспаления и различных паранеопластических синдромов вызваны секрецией опухолями цитокинов , гормонов , простагландинов и / или других агентов системного действия; они полностью исчезают после успешного лечения DDL.
Патология
Гистологическое появление опухолей DDL (см. 2 в внизу гистопатологии липосаркомы секции) варьирует в широких пределах , но наиболее часто демонстрирует особенности недифференцированных плеоморфных саркомы (которые являются опухоли густонаселенные с переменно по размеру и форме клеток , содержащих вариабельность размеры и форму ядра ) или саркомы веретенообразных клеток (опухоли, состоящие из веретенообразных клеток на фоне соединительной ткани ). Различные части опухолей DDL часто показывают различия во внешнем виде их фоновых соединительных тканей: эти ткани могут быть миксоидными (то есть состоящими из прозрачного, похожего на слизь вещества, которое при окрашивании с использованием стандартного метода окрашивания H&E выглядит более синим или фиолетовым, чем красный цвет нормальных тканей) или миксоколлагеновый (т. е. с высоким содержанием коллагеновых волокон на миксоидном фоне) и в ~ 5% случаев имеют области остеоида (см. рис. 1 ниже в разделе «Гистопатология липосарком») или хрящевого материала . В опухолях также наблюдаются большие различия в составе клеток. Например, до 10% опухолей DDL имеют области с гистопатологией ALT / WDL, а в редких случаях DDL есть области, содержащие менинготелиальные завитки плоских клеток.
Генетика
Неопластические клетки как в DDL, так и в ALT / WDL несут аналогичные небольшие дополнительные маркерные хромосомы (sSMC) и / или гигантские маркерные хромосомы, которые содержат дополнительные части q-плеча хромосомы 12 на полосах с 13 по 15. Эта хромосомная область включает два гена, связанных с развитием опухоли. , гены MDM2 и CDK4 . Наличие дополнительных копий этих двух генов и / или их избыточно продуцируемых белковых продуктов является высокочувствительным и специфическим индикатором того, что липоматозная опухоль представляет собой ALT / WDL или DDL, а не какой-либо другой тип липоматозной опухоли. Сверхэкспрессия MDM2 и CDK генов, и / или другой генетический материал в sSMCs или гигантских хромосом - маркеров, подозреваемых в содействии развитию и / или прогрессирования DDL, а также опухоли ALT / WDL. Другие гены в sMMC и гигантской маркерной хромосоме, которые также сверхэкспрессируются в неопластических клетках ALT / WDL и DDL, включают HMGA2 , CPM , YEATS4 и DDIT3 . Однако по сравнению с неопластическими клетками ALT / WDL неопластические клетки DDL: 1) экспрессируют более высокие уровни генов в двух аномальных хромосомах; это может способствовать переходу от ALT / WDL к DDL; и 2) более высокие уровни продуктов гена на длинном плече хромосомы 1 на полосе 32, на длинном плече хромосомы 6 на полосе 33 и, примерно в 25% случаев, на коротком плече хромосомы 1 на полосе 32.2, которая содержит Ген JUN (этот ген сверхэкспрессируется в DDL, но не в ALT / WDL). Поскольку продукт гена JUN , c-jun , ингибирует гибель клеток и способствует пролиферации клеток, его избыточная продукция может способствовать прогрессированию ALT / WDL до DDL и / или злокачественному образованию опухолевых клеток DDL. Профилирование экспрессии генов (то есть измерение экспрессии продуктов тысяч генов, производимых клетками, тканями или опухолями) показало, что дифференцировка клеток адипоцитов и метаболические пути в ALT / WDL активируются, в то время как пути клеточной пролиферации и реакции на повреждение ДНК активируются. в DDL.
Диагностика
Гистопатологические данные DDL часто недостаточно ясны, чтобы поставить точный диагноз. Однако диагноз DDL подтверждается у людей: опухоли которых содержат ALT / WDL, смешанные с гистологическими компонентами DDL; с историей наличия предыдущего ALT / WDL; или у которых имеется забрюшинная липосаркома (DDL составляет ~ 57% всех забрюшинных липосарком). Опухоли DDL редко (<1% случаев) проявляются как поверхностные опухоли кожи; почти в 5 раз реже, чем ALT / WDL, возникают в глазнице; и крайне редко встречаются у детей. Обнаружение амплификации MDM2 опухолевых клеток является золотым стандартом диагностики для отличия WDL от липом, диспластических липом, атипичных сарком с веретенообразными клетками, плеоморфных липом и солитарных фиброзных опухолей. С другой стороны, обнаружение в опухолевых клетках сверхэкспрессированного гена CDK4 или присутствие либо специфических sSMC, связанных с ALT / WDL, либо гигантской маркерной хромосомы, убедительно подтверждает диагноз DDL или ALT / WDL. Клиническая картина, гистопатология и различия генов (например, сверхэкспрессия гена cJUN опухолевыми клетками сильно способствует диагностике DDL по сравнению с ATL / WDL) между двумя последними формами липосаркомы обычно помогают различать их.
Лечение и прогноз
Полная хирургическая резекция обычно является рекомендуемым лечением первой линии для локализованных опухолей DDL. Однако новые исследования показывают, что пациенты с опухолями DDL, ограниченными конечностью или туловищем, и прогнозируемая 10-летняя общая выживаемость, связанная с опухолью, составляет 51% или меньше, имеют улучшенные результаты при добавлении химиотерапии (например, доксорубицин плюс ифосфамид ). их хирургические режимы. Для этих локализованных форм DDL также может быть рассмотрена периоперационная лучевая терапия в соответствии с рекомендациями Национальной комплексной сети рака .
Забрюшинная DDL является наиболее распространенной, хирургически неразрешимой и серьезной формой DDL: частота рецидивов составляет 66%, а общая пятилетняя выживаемость - 54%. Первичным вариантом лечения забрюшинной DDL является хирургическая резекция. Фаза III клиническое испытание нашли небольшое различие в результатах лучевой терапии с последующей хирургической резекции по сравнению с только хирургической резекции при лечении забрюшинного DDL. В других клинических испытаниях фазы III пациенты с DDL с недоступными забрюшинными и / или метастатическими опухолями получали химиотерапию первой линии, сравнивающую доксорубицин с доксорубицином плюс ифосфамид или доксорубицин с гемцитабином плюс доцетакселом . В других исследованиях также изучалась ценность различных режимов химиотерапии. В этих исследованиях часто обнаруживалась небольшая разница в общей продолжительности жизни при сравнении, но наблюдались некоторые улучшения выживаемости без прогрессирования заболевания и других клинических параметров. Основываясь на этих исследованиях, рекомендованной терапией первой линии для забрюшинных и других хирургически невозможных или метастатических опухолей DDL является лечение химиотерапией на основе антрациклинов или, в случае резистентных к опухолям или рецидивов, химиотерапия эрибулином . Обзор, проведенный в 2020 году, показал, что среднее время выживания для DDL низкого гистопатологического уровня и высокого гистопатологического уровня составляет 113 месяцев и 48 месяцев соответственно. Необходимы дальнейшие исследования, чтобы предоставить доказательства эффективности лучевой терапии, химиотерапии и новых методов лечения во всех разновидностях DDL.
Новые методы лечения
Несколько новых схем лечения DDL и более агрессивных или других проблемных случаев ALT / WDL в настоящее время проходят клинические испытания . В настоящее время проводится фаза II клинического исследования абемациклиба у пациентов с предварительным или нелеченым DDL. Предварительный анализ показал, что этот ингибитор продукта генов CDK4 и CDK6 , серин / треонин-специфичные ферменты протеинкиназы, обеспечивает увеличенное среднее время выживания без прогрессирования заболевания, составляющее 30,4 недели. Фаза III многоцентровое, рандомизированное, двойное слепое, плацебо - контролируемая клиническое исследование abemaciclib находится в активной фазе и в ближайшее время (как указано в июле 2021 года) начать вербовку 108 лиц с усовершенствованным, рецидивирующей и / или метастатической DDL. Исследование спонсируется Sarcoma Alliance for Research through Collaboration в сотрудничестве с Eli Lilly and Company . Рибоциклиб , также ингибитор генов CDK4 и CDK6 , в комбинации с ингибитором mTOR , эверолимусом, проходит II фазу клинических испытаний на лицах с продвинутой стадией DDL или лейомиосаркомой . Регистрационное исследование фазы III (т. Е. Большое подтверждающее исследование, предназначенное для установления приемлемого профиля пользы / безопасности для получения одобрения регулирующих органов для точно определенного показания) оценивает безопасность и эффективность миладеметана по сравнению с трабектином у пациентов с неоперабельным (т. Е. считается, что резекция вызывает неприемлемую заболеваемость или смертность) или метастатический DDL, который прогрессировал на 1 или более предшествующих системных терапиях, включая, по крайней мере, 1 терапию на основе антрациклинов. Спонсор, Rain Therapeutics Inc, в настоящее время набирает для исследования 160 человек. Другое клиническое испытание фазы III посвящено изучению ингибитора MDM2 миладеметана в сравнении с трабектином , блокатором онкогенного фактора транскрипции FUS-CHOP , при MDM2- избыточной экспрессии ALT / WDL и DDL. Миладеметан продемонстрировал управляемую токсичность и некоторую активность, приводящую к стабильному заболеванию и / или нескольким частичным ответам на DDL.
Миксоидная липосаркома
Презентация
Миксоидная липосаркома (MLS), которая включает тип липосарком, называемый круглоклеточной липосаркомой, составляет ~ 30% всех липосарком. Пик заболеваемости приходится на четвертое и пятое десятилетия жизни людей, причем в большинстве исследований преобладают мужчины. Хотя MLS редко встречается у детей и подростков, это самая распространенная форма липосаркомы, диагностируемая в этих возрастных группах. MLS обычно представляет собой большую (от 1 до 39 см; в среднем 12 см), подвижную, четко очерченную, безболезненную массу, которая развивалась от 1 недели до 15 лет до постановки диагноза. Опухоли MLS локализуются в глубоко расположенных мягких тканях бедер (65–80% случаев), голеней (10–15% случаев), забрюшинного пространства (8% случаев) и рук (5% случаев). Примерно в одной трети случаев эти опухоли метастазируют в другие участки мягких тканей (например, забрюшинное пространство, грудную клетку или другую конечность), скелетную кость и / или легкое. Люди могут иметь эти метастазы, особенно в кости; Было рекомендовано, чтобы пациенты были проверены на наличие метастазов в кости с помощью медицинских изображений , включая рентген , компьютерную томографию и / или магнитно-резонансную томографию .
Патология
Гистопатологический анализ MLS (см. Рис. 3 и 4 ниже в разделе «Гистопатология липосарком») выявляет клетки, разбросанные по миксоидной матрице (т. Е. Фон соединительной ткани, который кажется более синим или пурпурным, чем красный цвет нормальной соединительной ткани, когда эти ткани являются правильно подготовлен, окрашен H&E и просмотрен под микроскопом). Эти клетки представляют собой липобласты , некоторые из которых имеют форму кольца-печатки (форма, предполагающая, что клетка может быть неопластической), овальной или круглой формы. Опухоли MLS могут быть гиперклеточными и содержать сплошные слои круглых клеток, которые составляют не менее 5% всех клеток, или низкоклеточные, заселенные клетками с мягкими ядрами и <5% круглыми клетками на фоне изогнутых капилляров, напоминающих узор из проволочной сетки. Опухоли, содержащие не менее 5% круглых клеток, классифицируются как высокозлокачественные, в то время как опухоли с <5% круглых клеток классифицируются как низкосортные. Опухоли MLS высокой степени злокачественности обычно протекают более агрессивно, чем опухоли MLS низкой степени злокачественности.
Генетика
Опухолевые клетки MLS фактически определяются их экспрессией слитого гена FUS-DDIT3 (также называемого химерным геном ), который встречается в> 95% случаев, или слитого гена EWSR1-DDIT3 , который встречается в остальных <5% случаев. В ФУЗ-DDIT3 формы слитого гена , как в результате транслокации (называется T (12:16) (Q13: p11)) между сайтом в DDIT3 гена в полосе 12 хромосомы 12 в руку и д месте FUS гена в полоса 11 на коротком плече хромосомы 16 (также называемая р-плечом ). Слитый белок (называемый также химерный белок) продукт этого химерного онкогена гена, FUS-DDIT3, как известно, задержать созревание жировых клеток и способствуют неоплазии. EWSR1-DDIT3 слитый ген (называемый т (12; 22) (q13; q12)) результаты от транслокации EWSR1 гена , расположенного в полосе 12.2 на хромосоме 22 в д руку с DDIT2 гена. Продукт гибридного белка гена EWSR1-DDIT3 , как и гибридный белок FUS-DDIT3, способствует неоплазии. Несмотря на эти отношения генов слияния, необходимы дальнейшие исследования для определения их вклада в развитие и / или поддержание опухолей MLS.
Диагностика
Опухоли MLS низкой и средней степени злокачественности могут быть идентифицированы гистологически по их классической морфологии характерной сосудистой сети, разбросанной по миксоидной строме. Однако опухоли MLS высокой степени злокачественности бывает трудно отличить от других круглоклеточных новообразований, особенно опухолей MLS высокой степени злокачественности, которые состоят из диффузно-клеточной и / или чисто круглой клеточной морфологии до такой степени, чтобы скрыть этот классический сосудисто-миксоидный паттерн. Обнаружение реаранжировок гена DDIT3 с геном FUS или EWSR1 с помощью гибридизации in situ или иммуногистохимии или транскриптов слияния РНК этих генов с помощью полимеразных цепных реакций в реальном времени подтверждает диагноз высокой, а также неоднозначные случаи низкой или низкой степени злокачественности. опухоли MLS средней степени злокачественности.
Лечение и прогноз
MLS обычно лечится хирургической резекцией, но может потребоваться более радикальное вмешательство, например, ампутация конечности может потребоваться, когда сосудисто-нервный пучок конечности нарушен . Сообщается, что послеоперационный риск рецидива в течение 3 лет после операции составляет ~ 15%, если не вся опухоль удалена, и ~ 10%, когда удаление опухоли завершено. Добавление лучевой терапии к хирургической резекции улучшило местный контроль опухолей MLS и было рекомендовано для лечения неоперабельных и рецидивирующих MLS. Однако необходимы дальнейшие исследования, чтобы определить ценность лучевой терапии в лечении различных разновидностей MLS. Было обнаружено, что эффективными являются режимы химиотерапии с использованием ифосфамида , антрациклина, такого как даунорубицин , дакарбазин и / или трабектин : клиническое испытание фазы III показало, что время выживания без прогрессирования у пациентов с MLS, получавших трабектин или дакарбазин, составляло 5,6 и 1,5 месяца соответственно. В 2015 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов одобрило трабектин для использования при неоперабельных и метастатических липосаркомах.
В целом, 10-летняя выживаемость пациентов с MLS составила 77%, что значительно дольше, чем при других формах липосаркомы. По сравнению с MLS с низким риском, MLS с высоким риском (риск определяется содержанием круглых клеток опухоли и / или другими неблагоприятными прогностическими показателями) ассоциируется с повышенной частотой метастазирования и, следовательно, с более коротким временем выживания. Увеличение размера опухоли (≥ 10 см) тесно связано с MLS более высокого уровня и, следовательно, с более коротким временем выживания. Другие факторы, которые были связаны с неблагоприятными исходами при MLS, включают наличие некроза опухоли, возраст> 45 лет, сверхэкспрессию гена P53 и мужской пол. Круглоклеточная форма миксоидных липосарком также имеет относительно плохой прогноз: в различных ретроспективных обзорах миксоидная липосаркома обычно считалась низкослойной и, следовательно, относительно чувствительной к химиотерапии, тогда как миксоидная липосаркома высокой степени (т.е. круглоклеточная) имела более высокие показатели. метастазов, вел себя более агрессивно и плохо реагировал на химиотерапию.Однако важно отметить, что почти во всех случаях миксоидных липосарком у педиатрических пациентов прогноз был отличным.
Новые методы лечения
PPAR-γ - агонист (т.е. активатор), efatutazone, был изучен в небольшой фазе I испытании на лиц с различными поздних стадий злокачественных новообразований. Препарат вызывал заметную стойкую реакцию у человека с MLS, что позволяет предположить, что агонисты PPAR-γ могут быть полезны для лечения этого заболевания. В ходе клинического испытания II стадии, проведенного в Италии, изучается влияние трабектина и пиоглитазона (другого агониста PPAR-γ) на людей со стабильными опухолями MLS. Исследование состоит из двух последовательных этапов. На первом этапе изучается реакция пациентов, получавших минимум 4 цикла только трабектином. Если достигнута стабильная форма заболевания, на втором этапе будут изучены эффекты дальнейшего лечения пациентов с исходной реакцией на терапию комбинацией трабектина и пиоглитазона. Приближается к завершению клиническое испытание стадии II для оценки эффективности сиролимуса (ингибитор MTOR ; сиролимус также известен как рапамицин) плюс циклофосфамид (химиотерапевтический препарат) при метастатических или неоперабельных MLS. Клинические испытания фазы II включают набор пациентов для оценки синтилимаба (человеческое моноклональное антитело IgG4, направленное против белка 1 запрограммированной гибели клеток, расположенного на поверхности клеток) в сочетании с двумя химиотерапевтическими препаратами, доксорубицином и ифосфамидом , в качестве лечения первой линии мягкого тканевые саркомы, включая MLS.
Т-клетки были генетически сконструированы для нацеливания на антиген MAGE-A4, экспрессируемый на пептиде, содержащем HLA-A * 02 MAGE-A4, расположенном на поверхности неопластических клеток в некоторых типах опухолей. Эти сконструированные клетки (называемые ADP-A2M4-T-клетки) атаковали и убивали различные культивируемые раковые клетки человека, несущие этот антиген, и на клинической стадии 1 исследования уменьшили различные типы солидных опухолей у пациентов, опухоли которых содержали неопластические клетки, экспрессирующие этот антиген. В клиническое исследование фазы II в настоящее время набирают людей для изучения эффективности и безопасности Т-клеток ADP-A2M4 (созданных из собственных Т-клеток реципиента) у HLA-A * 02-поститивных пациентов с метастатическим или неоперабельным MSGE-4 на поздней стадии. -положительные опухоли MLS.
Плеоморфная липосаркома
Презентация
Плеоморфные липосаркомы (PLS), которые составляют от 5% до 10% всех случаев липосарком, являются быстрорастущими, обычно большими (> 5 см) и безболезненными, но очень злокачественными опухолями адипоцитов. Они возникают в основном у лиц старше 50 лет с преобладанием женщин. Опухоли PLS у детей встречаются редко. Опухоли PLS присутствуют в ноге или руке (65% случаев), забрюшинном пространстве или брюшной полости (15% случаев) или в редких случаях на стенке туловища, семенном канатике , областях головы и шеи, стенке грудной клетки, полости таза , легочной плевре , перикард и позвоночник. Эти опухоли обычно локализуются в глубоких мягких тканях, и только 25% случаев встречаются в подкожных тканях. Редкие случаи PLS были представлены у людей с синдромами Ли-Фраумени или Мюира-Торре , двумя наследственными генетическими заболеваниями, которые предрасполагают больных к развитию различных типов рака.
Патология
Гистопатология опухолей PLS часто состоит из участков, напоминающих миксоидную липосаркому, смешанных с участками, содержащими недифференцированные клетки. Эти опухоли являются гиперклеточными и содержат по крайней мере несколько липобластов различной формы с плеоморфными ядрами. Часто встречаются участки некроза , иногда присутствуют гигантские клетки, некоторые из которых являются многоядерными и / или содержат поглощенные нейтрофилы , а в некоторых клетках можно увидеть капли гиалина, а также разбросаны внеклеточно по всей опухоли. Недифференцированный компонент этих опухолей чаще всего состоит из веретеновидных клеток, при этом в 25% случаев обнаруживаются клетки с морфологией эпителиоидных клеток . Эти опухоли имеют по крайней мере некоторые очаги с гистопатологией, сходной с гистиоцитомами миксофибросаркомного типа высокой степени злокачественности , опухолью, ранее называвшейся злокачественной миксоидной фиброзной гистиоцитомой.
Генетика
Неопластические клетки PLS содержат различные генные и хромосомные аномалии: ген TP53 удален или мутирован в 17–60% случаев; RB1 ген удален в 60% случаев; и ген нейрофибромина 1 теряется из-за инактивации мутаций в 8% случаев или в более редких случаях из-за делеции вокруг его местоположения в полосе 11.2 на длинном плече хромосомы 12. Эти клетки также могут демонстрировать прирост генетического материала вокруг: полос 12 –15 на коротком плече хромосомы 5; полоса 21 на коротком плече хромосомы 1; и полоса 22 на длинном плече хромосомы 7. Изменения в количестве копий генов, вызванные этими аномалиями, аналогичны изменениям, наблюдаемым в гистиоцитомах типа миксофибросаркомы . Роль (и) этих изменений числа копий гена в продвижении PLS не определена. Таким образом, PLS отличается от других липосарком тем, что его неопластические клетки имеют сложный геном без характерных геномных изменений или идентифицируемых генов, которые вызывают заболевание. Обнаружение изменений в экспрессии ТР53, RB1 и Нейрофибромин 1 гены, а также другие, менее часто измененные гены в PLS (например , PIK3CA , тирозин-протеинкиназа Syk , PTK2B , EPHA5 и ERBB4 ), может помочь поддержать , но четко не определяют опухоль как PLS. Удлинение концов теломер хромосомы с помощью патологических механизмов, называемых альтернативным удлинением теломер, происходит в неопластических клетках примерно в 80% случаев PLS, но гораздо реже или не наблюдается при других четырех формах липосаркомы.
Диагностика
Диагноз PLS зависит от его проявления, гистопатологии и генетики. Гистопатология PLS часто очень напоминает гистопатологию миксофибросаркомы, но отличается от этой опухоли содержанием плеоморфных липобластов.
Лечение и прогноз
Радикальная хирургическая резекция - основное лечение PLS; это также важное паллиативное вмешательство для облегчения симптомов сдавления органов и тканей. Операция может потребовать удаления всего сдавленного органа, такого как почка или толстая кишка. Однако, независимо от этой операции, частота местных рецидивов очень высока. Не было доказано, что использование химиотерапии и / или лучевой терапии в сочетании с радикальным хирургическим вмешательством продлевает выживаемость и считается спорным вмешательством. National Comprehensive Cancer Network рекомендует лечение для людей с высоким риском локализованного ПЛСА по полной хирургической резекции, когда это возможно, в сочетании с лучевой терапией. Лица с метастатическим заболеванием получали химиотерапию (например, доксорубицин плюс ифосфамид или эрибулин ), аналогичную схемам, используемым для дедифференцированной липосаркомы (см. Выше раздел о лечении этого типа липосаркомы). Около 20% опухолей PLS метастазируют в отдаленные участки, в большинстве случаев распространенными из которых являются легкие (82% метастазов), печень (18% метастазов) и кость или поджелудочная железа (18% метастазов). Сообщается, что выживаемость PLS через 1, 3 и 5 лет составляет 93%, 75% и 29% соответственно. Опухоли, расположенные в центре туловища, размером более 10 см, глубоко расположенные или содержащие участки некроза, имеют худший прогноз.
Миксоидная плеоморфная липосаркома
Миксоидная плеоморфная липосаркома (первоначально называемая плеоморфной миксоидной липосаркомой) была впервые описана в большом исследовании липосарком в 2009 году. Первоначально рассматриваемый как возможный вариант миксоидной липосаркомы с плеоморфными особенностями, Всемирная организация здравоохранения (2020) классифицировала его как новую и отличную форму липосарком. Эта классификация была основана на выводах о том, что миксоидные плеоморфные липосаркомы, имея гистопатологические особенности, похожие на миксоидные липосаркомы, имели клинические и, что наиболее важно, критические генетические и молекулярные особенности, которые отличались от миксоидной, а также от трех других форм липосарком.
Презентация
Миксоидная плеоморфная липосаркома (MPL) - исключительно редкая и очень агрессивная форма липосарком, которая развивается у детей, подростков, молодых людей и, в более позднем исследовании, у людей старше 50 лет. Опухоли MPL представляют собой глубокие образования мягких тканей, которые часто расположены в средостении и, реже, конечностях, голове и шее, брюшной полости или туловище. По крайней мере, два случая MPL были представлены индивидуально с синдромом Ли-Фраумени , наследственным генетическим заболеванием, которое предрасполагает людей к развитию различных видов рака.
Патология
При гистопатологическом анализе опухоли MPL состоят из областей, напоминающих обычную миксоидную липосаркому; эти области, которые составляют 30–50% от общей площади опухоли, имеют обильный миксоидный матрикс, хорошо развитую капиллярную сосудистую сеть, мягкие клетки, имеющие округлую и / или слегка веретеновидную форму, вакуолизированные липобласты и многоядерные клетки, имеющие форму небольших цветы. Тем не менее, эти области также содержат множество высоко плеоморфных клеток, которые показывают большую степень увеличения и нерегулярности ядер, чем клетки, наблюдаемые в опухолях миксоидной липосаркомы. Другие области опухолей MPL более клеточные и состоят из быстро растущих и высоко плеоморфных липобластов .
Генетика
Неопластические клетки в MPL не экспрессируют слитые гены FUS-DDIT3 или EWSR1-DDIT3 , которые экспрессируются опухолевыми клетками в> 95% или <5% случаев миксоидной фибросаркомы, соответственно. Инактивация гена-супрессора опухоли RB1 из-за его делеции или патологического подавления обнаруживается во всех случаях MPL. Неопластические клетки MPL также обычно имеют другие изменения в своих хромосомах. Они могут показывать аномальные приросты в некотором генетическом материале, обычно обнаруживаемом на хромосомах 1, 6, 7, 8, 19, 21 и / или X, и потери генетического материала, обычно обнаруживаемые на хромосомах 2, 3, 4, 5, 10. , 11, 12, 13, 14, 15, 16, 17 и / или 22. генетический материал теряется в полосе 14 на длинном плече хромосомы 13 , включает в себя не только RP1 ген , но и RCBTB2 , DLEU1 , и ITM2B генов. Из-за его редкости и более позднего определения молекулярные характеристики и важность этих генетических аномалий еще предстоит полностью определить. Тем не менее, исследования показали, что потери в одном или нескольких генах RB1, RCBTB2, DLEU1 и ITM2B , но особенно в гене RP1 , могут участвовать в развитии и / или прогрессировании MPL.
Диагностика
Диагноз MPL зависит от клинического проявления опухоли, гистопатологического сходства с миксоидной липосаркомой и, что наиболее важно, отсутствия слитых генов FUS-DDIT3 sn EWSR1-DDIT3 в его неопластических клетках.
Лечение и прогноз
В то время как пациенты с MPL подвергались хирургической резекции для удаления опухолей, обзор 2021 года показал, что не было согласованных рекомендаций по стандарту лечения MPL в отношении режимов лучевой и химиотерапии (при использовании отдельно или в сочетании с хирургическим вмешательством) для лечение этих опухолей.
Гистопатология липосарком
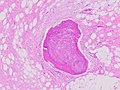
Рис.1 Микрофотография костеобразования при опухоли липосаркомы.

Рис. 2 Микрофотография дедифференцированной опухоли липосаркомы.

Рис. 3 Нижней мощности микрофотография из миксоидной опухоли липосаркомы

Рис. 4 Миксоидная микрофотография опухоли миксоидной липосаркомы в увеличенном разрешении.


.jpg)
.JPG)
Медицинская визуализация
Медицинское ультразвуковое исследование и магнитно-резонансная томография (МРТ) липосарком полезны и часто необходимы для определения их степени, хирургической доступности и связи с любыми наблюдаемыми дисфункциями органов. Поскольку ультразвуковое исследование обычно не позволяет отличить липосаркому от доброкачественной липомы, МРТ является предпочтительным исходным методом визуализации для получения доказанных доказательств в отношении определения этого отличия.

Рис. 5 Ультрасонография липосаркомы с участками с высоким эхом, отраженными от липоматозной матрицы, и с участками с низким эхом, отраженными от нелипоматозных областей.
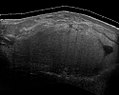
Рис. 6 Ультрасонография липосаркомы, имитирующей липому. Эта однородная высокоэхогенная масса выглядит так же, как липома.

Рис. 7 МРТ миксоидной липосаркомы высокой степени злокачественности в левой подмышечной области 40-летнего мужчины, выделенная белым цветом на этом горизонтальном срезе опухоли.



Общество и культура
Известные случаи
- Чад Браун (1961–2014), игрок в покер, умер от липосаркомы.
- Ричард Фейнман (1918–1988), физик-теоретик, умер после хирургической операции по борьбе с этим заболеванием.
- Роб Форд (1969–2016), бывший мэр Торонто и член городского совета Торонто, умер от плеоморфной липосаркомы.
- Хоки Гаджан (1959–2016 гг.), Бывший бегун от New Orleans Saints и комментатор по радио от команды, умер от липосаркомы.
- Чарли Дэвис (1986-), бывший футболист Филадельфийского союза футбольной высшей лиги, в 2016 году у него диагностировали липосаркому.
- Марк Стрэнд (1934-2014), бывший лауреат поэтессы США и лауреат Пулитцеровской премии, умер от липосаркомы.
Смотрите также
- Липома
- Wendy Walk , некоммерческая организация, миссия которой - сбор средств и повышение осведомленности о саркомах, в том числе липосаркомах.
использованная литература
внешние ссылки
| Классификация | |
|---|---|
| Внешние ресурсы |